SLiCE from Escherichia coli laboratory strains
![]() SLiCE (Seamless Ligation Cloning Extract) from Escherichia coli laboratory strains
SLiCE (Seamless Ligation Cloning Extract) from Escherichia coli laboratory strains
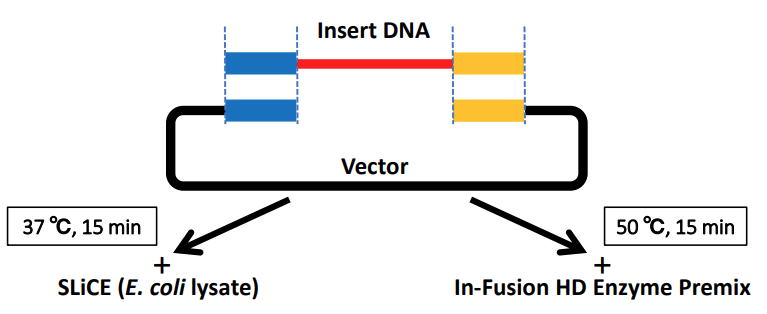
| 最近は、昔ながらの制限酵素を用いたベクター構築ではなく、制限酵素部位に依存しない様々なクローニング法を活用できます。ここで紹介するSLiCE
(Seamless Ligation Cloning Extract)法は、E. coli lysateの持つ内在性のシームレスクローニング活性をin vitroで用いて、制限酵素部位に依存せずPCR断片をベクターへクローニングする方法です(いわゆるseamless cloning)。このseamless
cloningに関連する試薬は、Gibson assembly kitやIn-Fusion kitなどの名称で様々なメーカーから商品化されていますが、その多くが高価で、資金の乏しい研究室では購入を躊躇しているかもしれません。そのような方に、ぜひ試していただきたいのがこのSLiCE法です。 |
||
 |
||
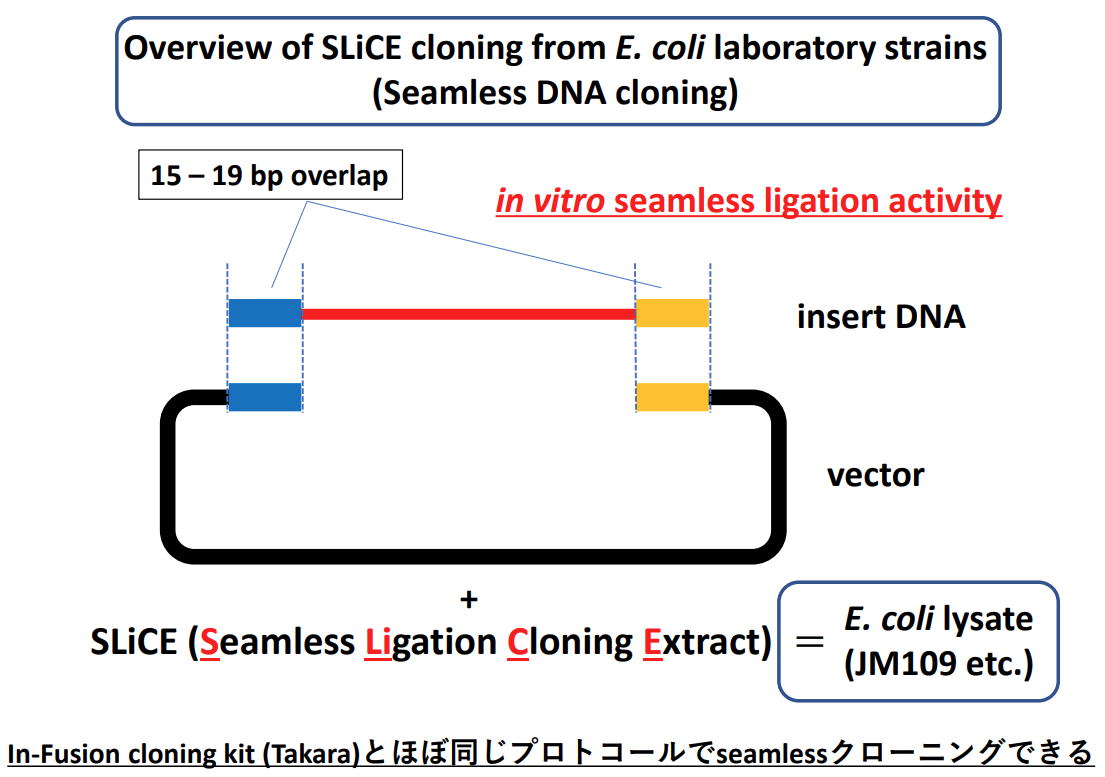
| 1. SLiCE from Escherichia coli laboratory strains この方法のオリジナルは、lambda prophage Red recombination systemを発現させたE. coli PPY株から、シームレスクローニング活性の高い抽出物を調製したSLiCE法にあります (ref.1)。この論文では、ファージシステムを発現していない通常のE. coli抽出物でも、効率は低いながらin vitro シームレスクローニング活性を示すことを報告していました。しかし、末端相同領域の短い15-20 bpでクローニングを行う場合、形質転換時のコロニー形成率が低すぎて、日常のPCRクローニングを高い効率で行うことはできませんでした (ref.1)。 私は、SLiCE法の汎用性を広げるため、研究室で通常用いられている一般のE. coli株を用いて、SLiCEの調製法と反応条件を最適化しました。その結果、このSLiCEが15-19 bpの短い末端相同領域の断片でも通常のDNAクローニングに十分な効率を持つことを示しました (ref.2)。これは、特別なE. coli株を使うことなく、研究室で使われているE. coli株から粗抽出液を調製するだけで、効率良くseamless cloningできることを示しています。大腸菌からのSLiCE調製は、CelLytic B Cell Lysis Reagent (Sigma, B7435)、または、 Triton X-100を含む緩衝液を使用します(ref.3, ref.4)。 |
||
 |
||
| 2. SLiCE-mediated PCR-based site-directed mutagenesis (SLiP-method) site-directed mutagenesisのひとつの方法としてQuikChange法が頻繁に用いられます。しかし、この方法はベクター領域をPCR増幅するため、PCR反応に長時間を要し、加えてベクター領域への意図しない変異導入の可能性も残ります。私は、この問題を解決するために、SLiCE法を応用し、簡単に点変異を導入する方法 (SLiCE-mediated PCR-based site-directed mutagenesis (SLiP-method)) も開発しました(ref. 2)。SLiP methodは、未精製PCR断片を2つ同時にベクターへ導入できるので、QuikChange法とほぼ同程度の簡便さでsite-directed mutagenesisが可能です。また、PCRによるベクターへの意図しない変異導入も排除できます。 |
||
| ---------------------------------------- | ||
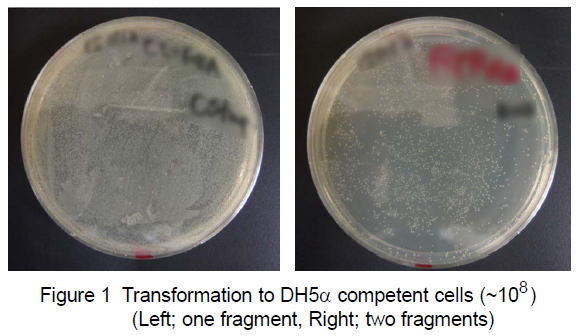
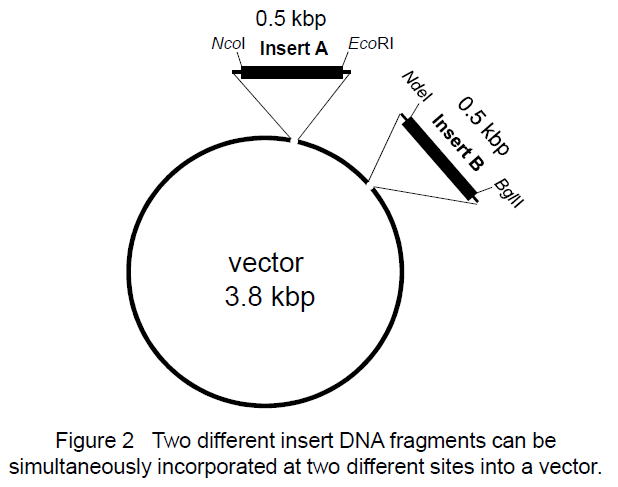
| 1.Preparation of SLiCE (from JM109) (ref. 5) JM109 stored at -80℃ ※DH5a, XL10-Gold, Mach1-T1Rなどでも可能だが、当研究室ではJM109を使用。 | 1mL LB is maintained at 37℃, usually for 3 h. | 50mL 2xYT is maintained at 37℃, usually for 4-5 h. Harvest the cells at OD = 2 to 3. ※If OD does not reach to 2-3, incubate overnight at 37℃. Next day, transfer 1mL overnight culture into another 50mL 2xYT and incubate at 37℃ for several hours. ※We measure the OD by UV-visible spectrophotometer V-650 (JASCO corp.). Centrifuge at 5,000xg for 10min at 4℃. Wash in 50mL ice-cold Milli-Q water. Centrifuge at 5,000xg for 5min at 4℃. 0.3-0.4 g of cells (wet weight) Resuspend gently in 1.2mL CelLytic B Cell Lysis Reagent (Sigma, B7435) (ref.2). ※Alternatively, buffers containing Triton X-100 can also be used (ref.3, ref.4). Incubate it for 10min at room temperature. ※Leave to stand, not rorate. Centrifuge at 20,000xg for 2min at 4℃. ※If you want to evaluate efficiency of protein extraction, you should check protein concentration of supernatant (ref.3). Place the supernatant on ice and add 1 volume of ice-cold 80% glycerol (v/v). Aliquot 40uL of each SLiCE extract into a 0.2mL-PCR tube. Snap-freeze in a bath of liquid nitrogen. Maintain this stock solution at -80℃ (for use, 40uL each extract in 0.2mL 8-strip PCR tube). ※SLiCEs can be stored at -80℃ for at least three years without significant loss of activity. ※96-well paper box for PCR-tube (0.2mLx8) is useful, when SLiCEs in 8-strip PCR tubes can be stored at -80℃. The 96-well papaer box for 0.2mLx8 PCR tube is here. ※For short-term storage, SLiCEs are stored at -20℃. ※SLiCEs can be stored at -20℃ for 2-3 months without significant loss of activity. 2.10 x SLiCE buffer(0.2um filtered), -20℃ stock 500mM Tris-HCl (pH7.5) 100mM MgCl2 10mM ATP 10mM DTT 3.Vector ・PCR amplified vector or restriction enzyme-digested vector。どちらでも可能。 ・多少のflanking sequenceは許容できる(ただし、効率は下がる)。 ・当研究室では、代表的な箇所を切断したベクターを調製し、精製後、ストックしている。 (多少の制限酵素部位のズレについては、この反応では問題にならないので、少し離れたところでも使用可。) 4.Insert DNA ・19bp overlap regionとなるようにプライマーを設計する(15bp以上でも可)。 ・正確性の高いDNAポリメラーゼを使用し、断片を増幅。 5.Standard protocol ・PCR (19bp overlap sequence) ・Purification of PCR fragments (by silica column) If multiple bands were amplified by PCR, the PCR fragments should be purified by agarose gel electrophoresis. ・vector(10-100ng) + insertDNA (insert:vector=1:1 to 3:1) ※Excess insert DNA inhibits the SLiCE reaction (ref. 4). ・SLiCE reaction InsertDNA VectorDNA 10xB 1uL SLiCE 1uL DDW up to 10uL 37℃, 10-60 min (Longtime incubation reduce colony formation rate.) ※A SLiCE reaction for 15 min results in enough cloning-efficiency, in many cases. 6.Rapid protocol ・PCR (19bp overlap sequence) ・vector(10-100ng) + unpurified insert DNA 1uL ・SLiCE reaction Unpurified insertDNA 1uL ※DpnI treatment efficiently reduces background colony formation. ※When a restriction enzyme-digested vector is used, DpnI should be inactivated for 15 min at 80°C. VectorDNA 10xB 1uL SLiCE 1uL DDW up to 10uL 37℃, 15min 7.SLiCE-mediated PCR-based site-directed mutagenesis (SLiP site-directed mutagenesis) (ref. 2, ref. 5) ・Design of mutation primers by PrimerX Condition1: melting temperature >78℃ Condition2: primer termination in G or C for QuikChange site-directed mutagenesis kit. ・変異領域を挟む二断片のPCR増幅 ・SLiCE for Rapid protocol Unpurified fragmentA 1uL Unpurified fragmentB 1uL ※DpnI treatment efficiently reduces background colony formation. ※When a restriction enzyme-digested vector is used, DpnI should be inactivated for 15 min at 80°C. VectorDNA 10xB 1uL SLiCE 1uL DDW up to 10uL 37℃, 15min (必要に応じて、PCR断片を精製し、Standard protocolでSLiCE反応) ---------------------------------------- SLiCE-related FAQ (Frequently Asked Questions) (2020/3/7改訂) これまで、多くの方々からSLiCE法について質問をいただきました。そのうち、代表的なものをFAQとしてまとめました。 Q1. SLiCEを調製するのに用いる大腸菌株は、どの菌株が良いですか。 A1. SLiCEを調製する大腸菌は、通常研究室で使用しているJM109, DH5a, XL10-Gold, Mach1-T1Rなど多くの大腸菌株を使用できます(ref.2)。どの菌株でも効率に大きな差はありませんが、通常、私たちはJM109から調製しています。 Q2. SLiCEを調製するのに適切なスケールを教えてください。 A2. 50 mL培地からSLiCEを調製すると、2000回以上のSLiCE反応を行うことができます。一度調製すれば、数年間は調製する必要がないと思われます。 ※通常、コンピテントセルは、100 mL培地から80本程度調製できます。一方、SLiCEは、50 mL培地で2000反応分以上も調製できます。 ※SLiCEを自家調製し、市販のシームレスクローニングキットの代わりにコンピテントセルを購入するのもよいかもしれません。 Q3. SLiCEの調製は、どの程度たいへんですか。 A3. 大腸菌からのSLiCEは、1日で簡単に調製できます。事前に準備するものは、液体培地、細胞破砕溶液(市販品、またはTris/Triton緩衝液)、MilliQ水、80%グリセロールの4つのみです。研究室でコンピテントセルを作成しているのであれば、それよりはるかに少ない労力で調製できます。 Q4. プロトコールに従って培養した場合、夜になっても大腸菌培養液がOD=2-3に達しません。どうしたら良いですか。 A4. そのまま、一晩培養してください。翌朝、増殖した培養液から1mLを採取し、新しい50mL培地に再度植菌してください。 Q5. 大腸菌を培養中にOD=3を超えてしまいました。どうしたら良いですか。 A5. 増殖した培養液から1mLを採取し、新しい50mL培地に再度植菌してください。 Q6. 調製したSLiCEは、長期保存できますか。 A6. -80℃で保存した場合、少なくとも5年間は安定です。長期保存には少量ずつ分注し、液体窒素凍結後、-80℃保存します(8連0.2mLPCRチューブを使用すると便利です)。使用する際には、1本ずつ取り出して、-20℃保存します。-20℃保存のSLiCEは2-3か月間安定に使用できます。私たちの研究室では、2か月経過したSLiCEは処分し、新しいSLiCEを-80℃から取り出すことにしています。温度変化を最小限に抑えるために、-20℃での保存には制限酵素保存に用いられる保冷ラック(Cooler rack)の使用をお薦めします。 これまでの経験では、-20℃に5ヶ月半保存したSLiCEでもクローニングできました(1断片のクローニング)。ただし、このときのクローニング効率は低いものでした。 Q7. 従来の制限酵素法によるベクター構築と比較して、効率は良いですか。 A7. 制限酵素処理したPCR断片をベクターに挿入する従来の制限酵素法と比較すると、SLiCEによるシームレスDNAクローニングは格段に効率の高い方法です。そのため、研究室の新人でも失敗がなくなりました。ただし、効率が良すぎて、プレート上に分離しないほど多数のコロニーが生えることがあるので注意が必要です(Figure 1)。 Q8. 市販のシームレスDNAクローニングキットの代替として使用することはできますか。 A8. 調製したSLiCEは、市販のシームレスDNAクローニングキットの代替法として十分に機能します(ref.4)。ただし、これまでのところ私たちの試した範囲では市販キットの効率を超えることはありません。市販キットよりもSLiCEの方が効率良い、という情報をいただくこともありますが詳細はわかりません。相同領域のDNA配列によって、効率が異なるのかもしれません。 Q9. ベクターとインサートの相同領域は、どれくらいの長さが適切ですか。 A9. SLiCE法で使用する際のインサートとベクターの相同領域長は、19-bpを推奨しています。これは、相同領域長の異なる断片で効率を比較した際、19-bpが最もクローニング効率が高かったためです(ref.2, Table 2)。ただし、その差はごくわずかであり、15-bp相同領域長でのクローニングでもまったく問題ありません(ref.3, ref.4)。最近のDNA合成は非常に安価ですので、保険としての+4-bpとお考えください。19-bpにすることでプライマーが長くなり到着が遅れるような場合には、私たちも迷わず相同領域長15-bpでプライマーを設計しています。 Q10. 挿入するベクターの調製法はどの方法が適切ですか。 A10. 挿入するベクターは、PCR増幅したベクター、制限酵素処理したベクターどちらでもかまいません(効率もほぼ同等)。また、多少のflanking regionなら、この反応は許容します(ref.2, Table 3)。当研究室では、代表的な制限酵素で切断したベクターを多めに調製しておき、そこへPCR断片を挿入しています(制限酵素部位の多少のズレは問題になりません)。ただし、flanking regionの長さに応じて、クローニング効率は低下します(ref.2, Table 3)。 Q11. PCR増幅した断片を精製する必要はありますか。 A11. 結論だけを言うと、未精製PCR断片でもSLiCE法でクローニングできます。ただし、精製DNA断片を使用したときよりもクローニング効率は低下します。各精製法によるクローニング効率を参考に、精製法を決定していただくのが良いと思います(ref.2, Table 4)。手間と効率のバランスを考えると、Gel&PCR精製キット(例, Nippon Genetics, FastGene Gel/PCR Extraction Kit)を使用するのが効果的です。このキットは、精製の手間に比して、ゲル電気泳動からの切り出しサンプルと同等のクローニング効率でした。 通常、私たちはPCR後のサンプルの1/10 vol.をゲル電気泳動によりPCR増幅を確認しつつ、並行して9/10 vol.をGel&PCR精製キットで精製し、SLiCE反応に用いています。 Q12. In-Fusionキットでは、Cloning EnhancerでPCR産物を処理すると、そのままIn-Fusion反応に使えて便利です。SLiCE法でも同様な簡易処理法はありますか。 A12. SLiCE法でも同様な簡易処理法があり、それがExoSAP-IT(Thermo Fisher Scientific)処理です(ref.2, Table 4)。Cloning Enhancerと同様に37℃-15min+80℃-15minの処理で、PCR産物をそのままSLiCE反応に使うことができます。鋳型DNAの持込みが多い場合には、DpnIを同時に加えることもできます。ただし、PCR産物のExoSAP-IT処理(30 min)は手軽ではありますが、その効果は限定的です(ref.2, Table 4)。一方、Gel&PCR精製キットによる精製は10-15分間程度で終了し、少々手間はかかりますが、ExoSAP-IT処理よりも短時間で処理でき、かつ一桁高い効率を示すためお薦めできます(ref.2, Table 4)。 Q13. クローニング効率(インサート陽性率)は、どれくらいですか。 A13. SLiCE法の場合、ほぼすべてのコロニーはインサートが挿入されたプラスミドを持つので、colony PCRでポジティブクローンをスクリーニングする必要はありません(ref.2; Table 1 and 2, and ref.3; Table S1)。ただし、インサート陽性率はインサートや相同配列部分の塩基配列にも依存しているようです。 Q14. SLiCE反応液で大腸菌形質転換を行う場合、SOC培地を使用した回復培養は必要ですか。 A14. 使用する抗生物質に関わらず、回復培養はSLiCE法の形質転換効率を向上させます。私たちは、形質転換時の回復培養を行うことを強くお薦めします。 Q15. コンピテントセルは、どの程度の性能が必要ですか。 A15. 通常、私たちは約107CFU程度のコンピテントセルを使用して、SLiCE法を行っています。一断片のクローニングには、106CFU程度のコンピテントセルがあれば十分です。 Q16. 複数DNA断片のクローニングはできますか。 A16. 効率の高い方法のため、PCR断片を2断片同時にベクターへ挿入できます(実験を始めたばかりの学部4年生でも)。ベクターの異なる2箇所へDNA断片を挿入する複雑な場合でも、2断片同時に挿入することができます(Figure 2)。何断片まで同時にクローニングできるかを試したことはありません。しかし、これまで3断片程度のクローニングは日常的に行ってきました。ただし、次の点に注意点してください。SLiCEは-20℃で2-3ヶ月程度安定ですが、効率は徐々に低下します。多断片クローニングの際には、-80℃ストックから取り出したばかりのSLiCEを用いることをお薦めします。 Q.17. SLiCEで小さなDNA断片をベクターにクローニングできません。SLiCEは、ある程度以上の大きさのDNA断片でないとクローニングできないのでしょうか。 A.17. 小さなDNA断片でもクローニングできます。これまでの経験では、SLiCE法で42 bpのDNA配列をベクターへ挿入できました。ただし、小さなDNA断片をクローニングする際、複数断片がタンデムにベクターへ挿入されることがあります。インサートDNA断片のモル比が高い場合、この現象は顕著になります。インサートDNA断片がタンデム挿入される場合、ベクターに対してインサートDNA量を減らしてください(i:vのモル比を調整)。反応の基本条件は、モル比でi : v =1:1 - 3:1です。 ※他のグループの報告によれば、SLiCE法で90 bpの挿入配列をベクターに挿入できたとのことです。 Q18. SLiCEには、大腸菌由来の夾雑物が混入していそうです。DNAは分解されませんか。 A18. SLiCEは、大腸菌の粗抽出物なので、DNA分解酵素も含まれているかもしれません。そのため、SLiCEの反応時間、反応後の処理には次のような注意を払っています。SLiCE反応を長時間行うことは避けます。通常、37℃15分で充分です。また、反応後のSLiCE反応溶液は、すぐに形質転換に用います。すぐに形質転換できない場合は、ただちに凍結保存します。 Q19. SLiCEの調製コストは、どの程度ですか。 A19. 大腸菌粗抽出液がhomologous recombination活性をもつので、コストは限りなくゼロです。計算したところ、培地、反応Bufferに用いる試薬、細胞溶解に用いる抽出液などすべて含めても、¥0.4 Yen / reactionでした(チューブ代が半分以上)(ref.2, ref.4, ref.5)。Ligaseを用いるLigationよりも低コストです。これなら、明日からどなたでもシームレスクローニングが可能です。 Q20. 最近、 in vivo E. coli cloning (iVEC)法という簡便な方法もあります。わざわざin vitroでSLiCE反応を行う利点はありますか。 A20. 酵母や大腸菌は、細胞内に内在性の末端相同配列組換え活性を持っています。その活性を利用したクローニング法を酵母ではGap repair cloningと呼び、酵母細胞内で効率よくベクター構築を行えるようです。酵母の扱いに慣れている方は、良いクローニング法かもしれません(酵母の増殖速度が遅いことを除けば)。最近は、大腸菌でもPCR断片とベクターをin vitroで結合させることなく、形質転換するだけで大腸菌内でベクター構築できるとの報告があり、実際にできるようです。ただし、大腸菌のiVEC法の効率をSLiCE法と比較すると、SLiCE法は、iVEC法より1~2桁高い効率を示しました(ref.2 Table 1, "JM109" vs "no extract", ref.6)。方法の選択に関しては、37℃15分間のin vitro反応を余計な時間と考えるのか、クローニング効率を優先するのか、などを考慮のうえ、決定するのが良いでしょう。私たちは、37℃15分間の反応を行うことで格段にクロ-ニング効率が向上するため、SLiCE法を好んで使用しています。 Q21. QuikChange法とSLiP法によるsite-directed mutagenesisにはどのような違いがありますか、また、SLiP法を使用するメリットは何でしょうか。 A21. QuikChange法は、プラスミド全体をPCR増幅し、特定部位に変異を導入する方法です。これに対して、SLiP法はインサートのみをPCR増幅すれば良いので、DNA断片の増幅を短時間で行うことができます。また、増幅したPCR断片を精製することなく2断片を同時にベクターへ挿入できるので、QuikChange法とほぼ同じ手間で変異体作成できます。これに加えて、SLiP法では制限酵素処理したベクターを用いると、PCRによるベクター領域への意図しない変異導入を排除できます。 Q22. SLiCE法でクローニングする場合のおおよその時間を教えてください。 A22. 私たちの通常のタイムスケジュールを紹介します(急ぐ場合、精製法などを簡易法に変更します)。 Day1 14:00 Primer到着 14:30 PCR 16:30 PCR増幅の確認、およびDNA断片の精製 1/10 vol. アガロースゲル電気泳動による増幅の確認。 9/10 vol. FastGene Gel/PCR Extraction Kit (Nippon Genetics)精製。 ※ゲル電気泳動確認とDNA断片精製は並行して進める。 17:00 SLiCE (37℃, 15 min) ※ベクターは、あらかじめ制限酵素法で切断したものをストックしている。 17:30 Transformation 19:00 Plating on LB plate. ※PCR後のゲル電気泳動でPCR増幅断片が検出できない場合でも、次のステップへ進んでいます。 ※SLiCE法はクローニング効率が高いので、1/10 vol.でPCR増幅断片が検出できない場合でも、クローニン グできる例が数多くあります。 Day2 9:00 スクリーニング (Colony-PCR or Plasmid prep. from 2 mL LB medium) |
||
| ref.1) | Zhang, Y. et al., | Nucleic Acids Res. vol.40, e55 (2012). |
| ref.2) | Motohashi, K., | BMC Biotechnol. vol.15, 47 (2015). |
| ref.3) | Okegawa, Y. and Motohashi, K., | Anal. Biochem. vol.486, 51-53 (2015) |
| ref.4) | Okegawa, Y. and Motohashi, K., | Biochem. Biophys. Rep. vol.4, 148-151 (2015) |
| ref.5) | Motohashi, K., | Methods Mol. Biol. vol.1498, 349-357 (2017) |
| ref.6) | Motohashi, K., | Biochem. Biophys. Rep. vol.9, 310-315 (2017) |
| ---------------------------------------- 詳細は、以下の文献を参照ください。 実験医学2016年9月号 (vol.34, No14)の「クローズアップ実験法」に、SLiCE法に関する解説記事が掲載されました。 この方法を用いて実験され、論文作成する場合には、下記論文の引用をぜひお願いいたします(代表的論文: ref. 2, 3, 4, 5)。 また、生物工学会誌 2018年1月号 (vol.96, No1)の「続・生物工学基礎講座-バイオよもやま話-」に、SLiCE法をはじめとするシームレスクローニング法についての解説記事が掲載されました。こちらもご覧ください。 |
||
| ref.2 | Ken Motohashi | |
| "A simple and efficient seamless DNA cloning method using SLiCE from
Escherichia coli laboratory strains and its application to SLiP site-directed mutagenesis" BMC Biotechnol. 15, 47 (2015) |
||
| ref.3 | Yuki Okegawa and Ken Motohashi | |
| "Evaluation of seamless ligation cloning extract (SLiCE) preparation
methods from an Escherichia coli laboratory strain" Anal. Biochem. 486, 51-53 (2015) |
||
| ref.4 | Yuki Okegawa and Ken Motohashi | |
| "A simple and ultra-low cost homemade seamless ligation cloning extract
(SLiCE) as an alternative to a commercially available seamless DNA cloning
kit" Biochem. Biophys. Rep. 4, 148-151 (2015) |
||
| ref.5 | Ken Motohashi | |
| "Seamless Ligation Cloning Extract (SLiCE) method using cell lysates
from laboratory Escherichia coli strains and its application to SLiP site-directed mutagenesis" Methods Mol. Biol. 1498, 349-357 (2017) |
||
| ref.6 | Ken Motohashi | |
| "Evaluation of the efficient and utility of recombinant enzyme-free
seamless DNA cloning methods" Biochem. Biophys. Rep. 9, 310-315 (2017) |
||
  |
||
| SLiCE from Escherichia coli laboratory strains (in English) | ||
 |
||||
| Return to "Technical tips" | ||||
| E-mail: |