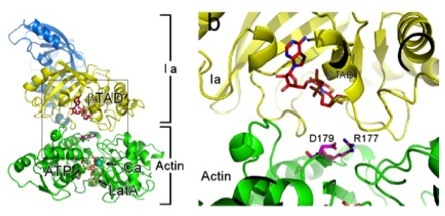
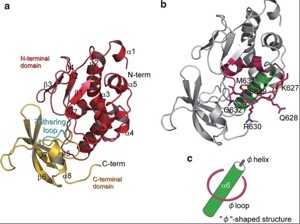
Protein Structural Biology
Faculty of life Science
*
Kyoto Sangyo University
**