Å@1. Functional analysis of collagen-specific molecular chaperone, Hsp47 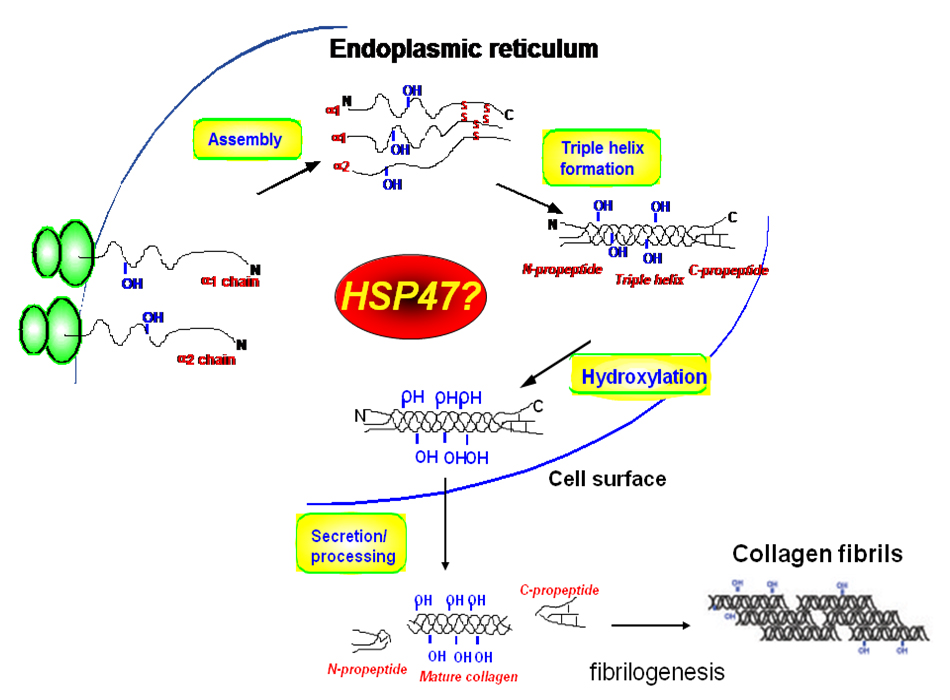
|
|
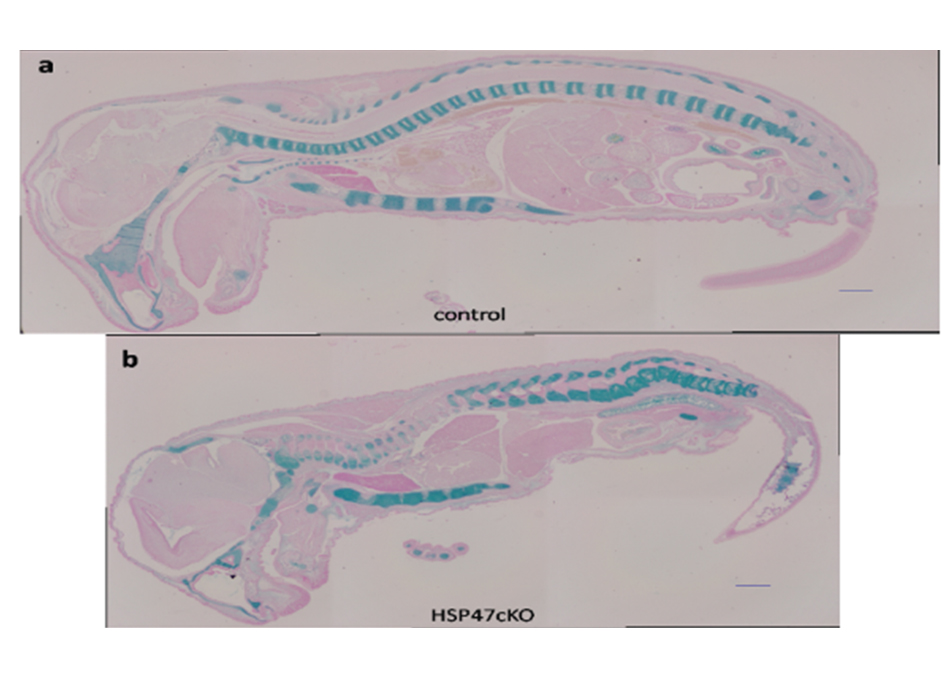
Å@We found and cloned the gene of a stress protein HSP47 which resides in the endoplasmic reticulum (ER) acting as a collagen-specific molecular chaperone in the pathway of collagen biosynthesis, processing and secretion. HSP47 specifically and transiently binds to various types of collagen in the ER. In addition to the binding specificity to collagen, the expression of HSP47 is always closely correlated with those of collagens during the normal development of mouse embryo as well as in the pathophysiological conditions including liver and renal fibrosis. Å@We already succeeded in making knockout mice lacking hsp47 gene, which resulted in causing the embryonic lethality at 10.5 dpc in hsp47-/- homozygotic mice. In these homozygotic mice, the maturation of type I collagen was abnormal and the immature form of procollagen accumulated in the tissues. Using hsp47-/- ES cells, we found that type IV collagen secreted from hsp47-null cells could not form correct triple helices and the basement membrane was not formed in the embryoid bodies from those cells. We also observed the impairment of basement membrane formation in mouse embryos, thus these findings reveal that the knockout of a chaperone protein HSP47 causes the abnormality in molecular maturation of its substrate, and HSP47 is essential for mouse normal development. In those knockout mice, type IV collagen was observed to accumulate in the ER causing an ER stress, and apoptosis was also observed in those embryos after 10.5 dpc. Those procollagens accumulated in the ER of the Hsp47 knockout cells were eliminated by autophagy, not by ER-associated degradation. Å@We also established conditional knockout mice with LoxP-Hsp47 gene and after crossing the mice with Cre gene under type II collagen-promoter, we observed the mice with sever cartilage and bone formation with abnormal molecular maturation of type II collagen. |
Å@2. Funtional analysis of a mammalian ER quality control and ER-associated degradation 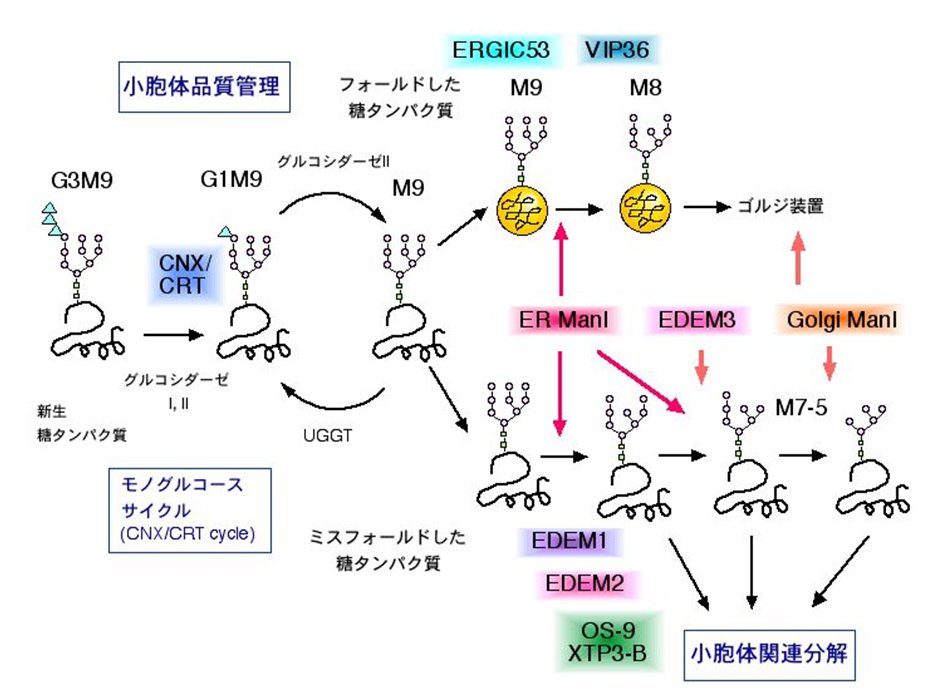
|
|
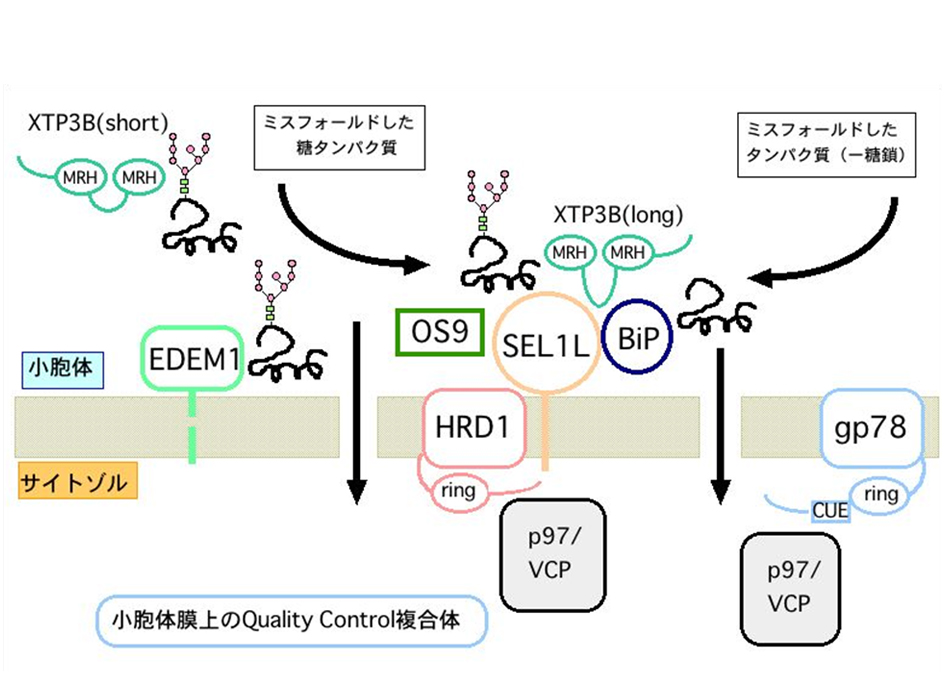
Å@We are working on the molecular mechanism of ERQC (ER quality control) and ERAD (ER-associated degradation). Many works have clarified the importance of ERAD of misfolded proteins and the disruption of ERQC in genetic diseases and neurodegenerative disorders. Since most of the proteins synthesized in the ER are N-glycosylated, ERQC of glycoproteins are regulated by the processing of the N-glycans and the recognition of specific N-glycans by the lectins. Recently, we have cloned two mammalian ER lectins OS-9 and XTP3-B. We have clarified the oligosaccharide structures that OS-9 lectin domain recognizes, and have shown that EDEM3 is capable of presenting this glycan signal for degradation by processing specific mannoses in vivo. We are now analyzing the oligosaccharide structures that XTP3-B lectin domains identify. Furthermore, we are analyzing the association and function of these lectins with a membrane-embedded ubiquitin ligase complex HRD1-SEL1L. The functional analysis of mammalian EDEM family proteins are also in progress. |
Å@3. ER-asociated degradation of misfolded proteins by EDEM-ERdj5 system 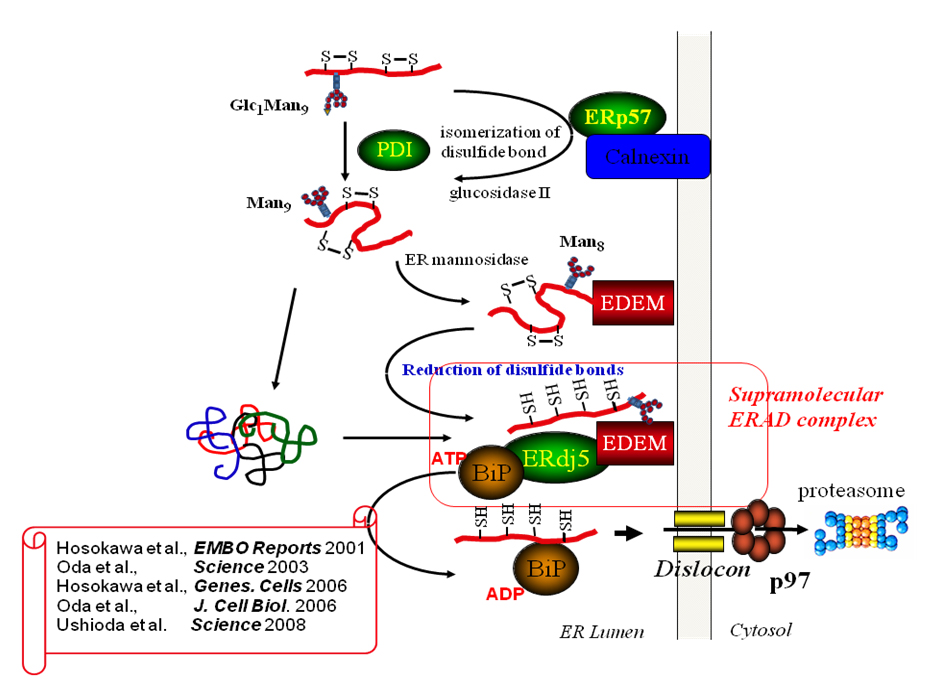 Å@We are working on the molecular mechanism of protein quality control by oxidoreductases in the ER. We identified that an ER disulfide reductase, ERdj5 promotes ERAD by cleaving of intermolecular disulfide bonds of misfolded proteins and preventing their oligomer or aggregate formation. ERdj5 interacts with EDEM, which recognizes misfolded proteins via their N-glycan portions, and a molecular chaperone, BiP. This ternary complex composing with ERdj5, EDEM and BiP accelerates the ERAD. After domain analysis of ERdj5, we found that EDEM interacts with ERdj5 via the C-terminal thioredoxin-like domain cluster and recruits misfolded glycoproteins to ERdj5 and thioredoxin-like domains at the C-terminal cluster are involved in cleaving the intermolecular disulfide bonds of misfolded proteins. BiP associates with ERdj5 through the J-domain in the N-terminus, and captures the reduced misfolded proteins until they are transferred to the dislocation channel.
Å@We are working on the molecular mechanism of protein quality control by oxidoreductases in the ER. We identified that an ER disulfide reductase, ERdj5 promotes ERAD by cleaving of intermolecular disulfide bonds of misfolded proteins and preventing their oligomer or aggregate formation. ERdj5 interacts with EDEM, which recognizes misfolded proteins via their N-glycan portions, and a molecular chaperone, BiP. This ternary complex composing with ERdj5, EDEM and BiP accelerates the ERAD. After domain analysis of ERdj5, we found that EDEM interacts with ERdj5 via the C-terminal thioredoxin-like domain cluster and recruits misfolded glycoproteins to ERdj5 and thioredoxin-like domains at the C-terminal cluster are involved in cleaving the intermolecular disulfide bonds of misfolded proteins. BiP associates with ERdj5 through the J-domain in the N-terminus, and captures the reduced misfolded proteins until they are transferred to the dislocation channel.Å@We have also established the interaction networks among around 20 ER oxidoreductases by co-immunoprecipitation and following LC/MS analysis to elucidate redox cascades in the ER. Based on the analysis, we have found that the redox complex composed of an ER oxidase, Ero1L and an ER oxidoreductase, PDI makes a regulatory hub complex, which oxidizes other ER oxidoreductases efficiently. We have also found that an ER oxidoreductase, TMX4, which is localized on the ER membrane, interacts with a lectin-like chaperone, calnexin and an ER oxidoreductase, ERp57, both of which are involved in the glycoprotein oxidative folding processes. |
 TOP
TOP